

66 Participants from 9 Nations

Joined Together To Form 9 Teams
For A 72h BioHackathon


Development
Each team was tasked with solving a unique problem put forward by 'team leads'. This required a diverse set of tools being used in the development of novel scientific solutions.

Collaboration
Individuals on each team contributed their unique skill-sets. Participants overwhelmingly described the experience as highly collaborative, fun and enlightening.

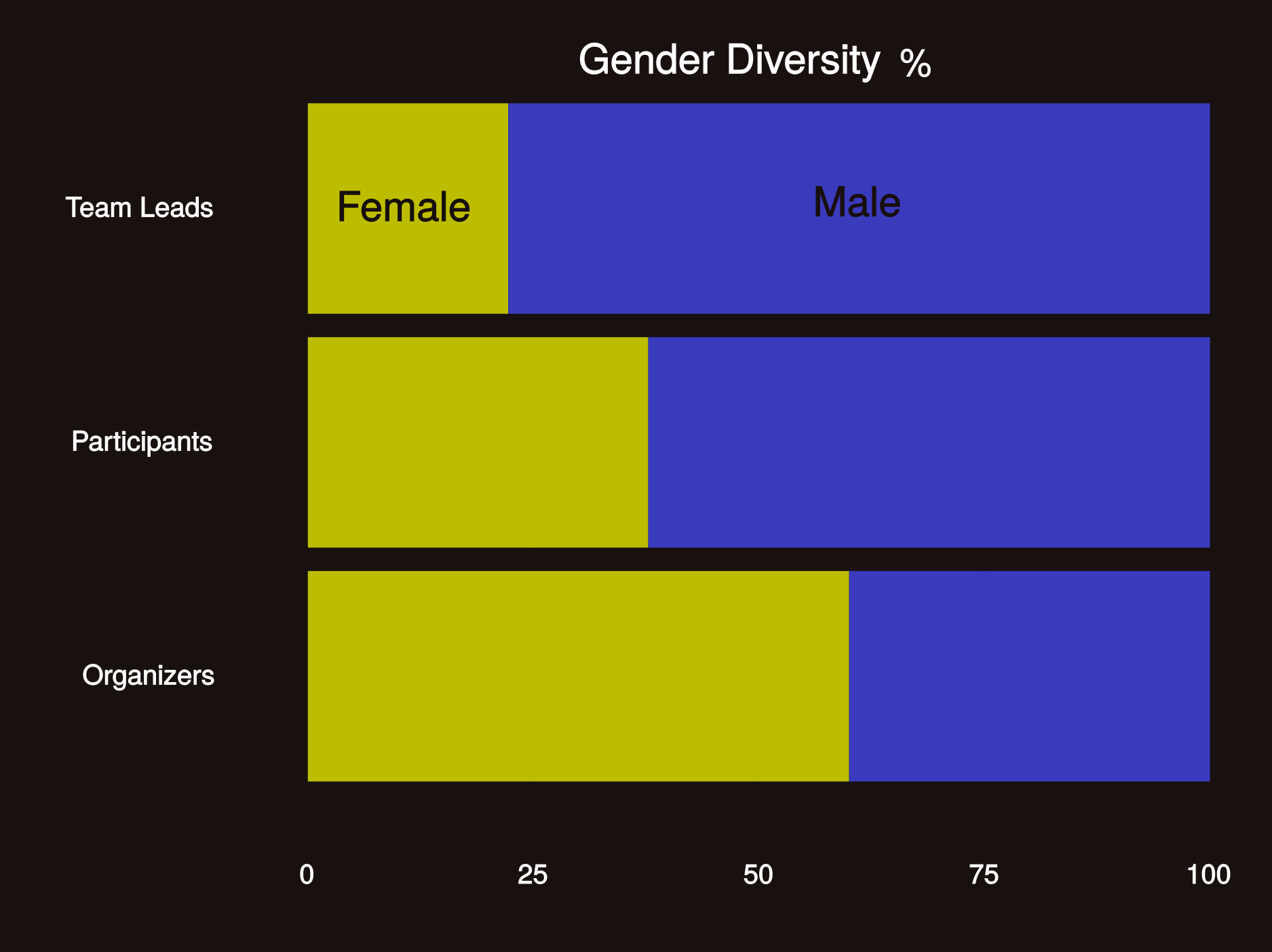
Share in the #hackseq16 experience
Read the hackseq16 Meeting Report on F1000
Project Abstracts
click to explore
+VASCO: Visualization App for Single Cell explOration {Winning Team}
Jean-Christophe Berube, Ogan Mancarci, Erin Marshall, Edward Mason, Celia Siu, Ben Weisburd, Shing Hei Zhan, Grace X.Y. Zheng*
*project lead
Abstract
Characterizing the transcriptome of individual cells is fundamental to understanding complex biological systems such as the nervous system, the immune system and cancer. Single cell RNA sequencing (scRNA-seq) technologies can profile transcriptomes of single cells. Recent development in scRNA-seq technologies has enabled profiling of thousands of cells at a time. However, existing analysis tools are designed for datasets with much lower cell numbers, and for people with a programming background, limiting their adoption. There is a strong need for an intuitive user interface to facilitate real-time data visualization and exploration by scientists to accelerate the discovery cycle of scRNA-seq analysis.
We have developed a visualization application, Visualization Application for Single Cell Overview (VASCO), that can be used for scRNA-seq data from thousands of cells in real time. It takes the gene-cell matrix and the cell clustering result as input. Users can visualize the cells using t-distributed stochastic neighbour embedding (t-SNE) plots. They can explore the expression pattern of specific genes. In addition, they can investigate the identity of cell clusters by examining genes that are specific to a cluster.
The application uses the plotly tool with Shiny to enable interaction over a web browser (currently tested on Firefox and Chrome). The source code is available on https://github.com/hackseq/vasco. All plots can be saved and tables exported. We have tested single cell datasets consisting of thousands of cells, but the application can be extended to support the interactive analysis of tens of thousands of cells.
| Video Presentation | Project Github |
+XYalign: Hacking sex chromosome variation
Madeline Couse, Bruno Grande, Eric Karlins, Tanya Phung, Phillip Richmond, Timothy H. Webster, Whitney Whitford, Melissa A. Wilson Sayres*
*project lead
Abstract
Sex chromosome copy number variations are currently estimated to be as common as 1/400 in the human population. Violations in typical ploidy will affect estimates of genome diversity and variation calling that is required in most clinical genomic studies. Further, mis-alignment of reads between the X and Y chromosomes will affect variant calling. Here we propose a new tool, XYalign, to quickly infer sex chromosome ploidy in NGS data (DNA and RNA), to remap reads based on inferred sex chromosome complement of the individual, and to output quality, depth, and allele-balance across the sex chromosomes.
| Video Presentation | Project Github | Slides |
+ParetoParrot: Design a tool to optimize the parameters of command line software
Craig Glastonbury, Daisie Huang, Hamid Younesy, Jasleen Grewal, Laura Gutierrez Funderburk, Lisa Bang, Shaun Jackman*, Veera Manikandan Rajagopal, Y. Brian Lee
*project lead
Project Goal
Given a command line tool with a number of parameters and a target metric to be optimized, I want a tool that will run the program and find the values for those parameters that maximizes some target metric. My particular use case is genome sequence assembly, which often has a variety of parameters related to expected coverage of the reads and heuristics to remove read errors and collapse heterozygous variation. When I tackle that optimization, the process is manual and tedious: submitting jobs to a scheduler, rerunning failed jobs, inspecting outputs, tweaking parameters, and repeating. I want to design and implement a tool to automate that process and generate a report of the result.
| Video Presentation | Project Github | Slides |
+BaklavaWGS: Pseudo-WGS variant calling for common cell types aggregating ChIP-seq, RNA-seq and DHS from ENCODE and Roadmap Epigenomics data
Carolyn Ch'ng, David Brazel, Karthigayini Sivaprakasam, Jill Moore, Shobhana Sekar, Stephen Kan, Jing Yun Alice Zhu, Ka-Kyung Kim, Luca Pinello*
*project lead
Abstract
Recent progress in sequencing technologies have led to a massive production of epigenetic datasets for many cell lines, thanks mainly to big consortia like ENCODE. This resource has helped tremendously to understand the role of non-coding variants and to prioritize the search of putative causal SNPs through cell-type-specific functional regions such as active or repressed promoters, enhancers, insulators, repressed or open chromatin. On the other hand we still miss genotype information for many cell types profiled limiting the power of allele specific alignments and further analysis that require the right reference genome.To fill this gap we propose a novel pipeline called baklavaWGS. This pipeline recovers genotype information such as SNPs for common cell types aggregating ChIP-seq, RNA-seq, DHS and other sequencing data already produced and available from consortia like ENCODE and Roadmap Epigenomics. Although single assays for each cell line don't have enough coverage in many regions of the genome, aggregating sequencing information for all the available assays for each cell type provides enough power to recover variants with high confidence.We evaluated the quality of our approach using a common cell line (NA12878) for which benchmark data was available from the Genome in a Bottle project. This effort will provide .VCF files for many cell types ready to use for the community, mining already available and public data.
| Website | Video Presentation | Project Github | Slides |
+A framework to evaluate profiles from DNA-binding site collections represented in peak sequences from ChIP-Seq assays.
Fotis Tsetsos, Kieran O'Neill, Shreejoy Tripathy, Manuel Belmadani*
*project lead
Project Goal
Our challenge here is to present a software framework allowing users to evaluate representation of a given motif (pattern) in a set of DNA sequences. These sequences are "peaks" from a ChIP-seq assay. In general, these peaks can represent the approximate location (site) where a transcription factor (TF), which is type of protein, will bind to regulate transcription of a gene. These are known as Transcription Factor Binding Sites.
On one part, we have repositories of ChIP-seq assays. You can expect to find:
1) The peaks (encoded by a list of tab-separated values as .bed file). The .bed file contains a chromosome, start location and end location for the peak.
2) The target of the experiment, representing the TF of interest.
3) Additional metadata, such as the tissue/cell line used in the assay, the organism, the experiment authors etc.
| Video Presentation | Project Github | Slides |
+Selection of tag SNPs for an African SNP array by LD and haplotype based methods
Ayton Meintjes, Scott Hazelhurst, Vincent Montoya, Marcia MacDonald, Jocelyn Lee, Dan Fornika, Brian Lee, Austin Reynolds, Tommy Carstensen*
*project lead
Abstract
Developing a cost-efficient and representative genotype array with SNPs that provide good coverage across the African continent is key to conducting large-scale medical genetic studies in Africa. The great amount of diversity across Africa has not previously been captured on any commercial SNP array to date. Significant new sequencing projects of African populations are generating rich sources of diversity, which can be used for chip design. However, none of the existing tag SNP selection algorithms for designing SNP arrays are geared towards handling WGS data efficiently. Our challenge was to (1) design an algorithm to quickly identify tag SNPs from WGS data and (2) apply this algorithm to a large African WGS dataset to produce a list of candidate SNPs for a commercial array. Our algorithm combines existing imputation methods with pairwise LD SNP tagging to identify candidate SNPs. It accepts standard Variant Call Format (VCF) files as input, and produces VCF files as output, which aids integration into extended analysis pipelines. By developing an efficient tag SNP selection algorithm that accepts next-generation sequence data, we hope to facilitate the continued improvement of whole-genome SNP assays for genetically diverse populations as new sequence data becomes available.
| Video Presentation | Project Github | Slides |
+Somatic Mutation from Separated Haplotypes (SMUSH)
Amanjeev Sethi, Eric Zhao, Hua Ling, Patrick Marks*, Peng Zhang, Samantha Kohli
*project lead
Abstract
Currently, accurate somatic mutation calling relies upon comparison of tumor to matched normal. The matched normal enables filtering of germline variants and differentiation of low frequency somatic mutations from sequence noise/errors. However, matched normal tissue is often unavailable. In this study, we investigate whether phasing information from linked reads can help differentiate somatic variants from germline alterations and sequencing errors.
Whole exome sequencing of the HCC1954 BrCa cell line was performed at 0%, 25%, 50%, 75% and 100% tumor purity. Library construction was performed using 10x genomics linked reads. Samples were sequenced at mean coverage of 200x. Sequence alignment and variant calling were performed using longranger and FreeBayes. Read counts of reference and alternate alleles were obtained for each haplotype after excluding reads with mapping quality < 30 to calculate variant allele fractions (VAF).
To differentiate between wild type, germline variants, and somatic mutations, we devised the SMUSH algorithm. Haplotype-specific VAFs were modeled as binomial random processes. model selection was performed using a maximum-likelihood estimate accounting for haplotype phasing, mutant read capture rates, and sequencing error.
To benchmark the accuracy of model selection, 31 somatic variants validated by comparison of 0% and 100% tumor content samples were manually reviewed in IGV and selected as a testing subset. Employing the SMUSH model on this subset correctly identified all variants as somatic in the 25-100% samples and correctly identified 30/31 variants as wild-type in the 0% sample.
| Video Presentation | Project Github | Slides |
+MetaGenius
Erik Gafni, Dan Kvitek, Jake Lever and Michael Schnall-Levin*
*project lead
Abstract
The analysis of short-read derived shotgun metagenomic data presents substantial challenges. While the reads can be assembled into short contiguous segments, the presence of homologous sequences within and among the genomes of the different species highly limits the ability to assemble these segments into anything close to full genomes. Here, we build a prototype assembler that uses 10x Genomics’ Linked-Read data to assemble shotgun metagenomic samples, and apply it to a dataset consisting of DNA derived from a mixture of 5 different bacterial species. Our assembler proceeds in multiple steps. First, it builds an initial assembly using the short-read data alone. Next, contigs from this initialy assembly are extended in a barcode-aware manner, and the relative localization of the contigs on the genomes are inferred by the fraction of barcodes shared between 2 contigs. A representative set of contigs that are inferred to be well-separated are then used to recruit reads by their barcodes and locally reassemble all reads from that region. This results in a set of contigs that are ~20-fold longer than the initial short-read derived contigs. These final contigs are scaffolded by both read-pairs and barcode information into very large scaffolds. Analysis of these scaffolds suggests that local barcode-based reassembly will be able to fill-in contig breaks within each scaffold. Our prototype suggests that with Linked-Reads it hould be possible to obtain highly complete genome assemblies from metagenomic samples.
| Video Presentation | Project Github |
+mICP: Metagenomic indicator contig predictor
Ben Busby*, Justin Chu, Jessica Hardwicke, Sean La and Feng Xu.
*project lead
Abstract
With the advent of affordable sequencing technology there has been a breakthrough in recent years of microbiome diversity studies. Many of these are solely on limited and often biased 16S rRNA amplicons. With the increase of publicly available, whole metagenomic datasets the scope of environmental classification can be broadened to include larger sequence contigs, including microbial “dark matter”. Here we present mICP, a novel strategy to predict indicator contigs for metagenomic datasets. To predict an initial set of indicator contigs, we used long reads (PacBio) from similar phenotypic (infant gut microbiome) datasets, and mapped short reads (Illumina) from divisible phenotypic groups (male vs female and vaginal vs caesarean birth) to these ‘contigs’, and set thresholds for coverage continuity and depth to define indicator contigs.
| Video Presentation | Project Github |